Last updated: July 14, 2026
Software as a Medical Device (SaMD) is software intended to be used for one or more medical purposes that performs those purposes without being part of a hardware medical device. It runs on general-purpose platforms such as cloud servers, smartphones, or hospital workstations. For cardiology practices, this definition directly captures cloud-based CIED data aggregation platforms and chronic-condition RPM tools that analyze physiological data and generate clinical alerts.
The FDA applies this definition under Section 201(h) of the Federal Food, Drug, and Cosmetic Act. A platform that ingests multi-OEM CIED transmissions, applies AI-driven alert triage, and produces clinical recommendations is almost certainly regulated as a medical device in its own right, regardless of whether it connects to implanted hardware.
See how Rhythm360 is built for FDA-aligned cardiac remote monitoring by requesting a demo.
The IMDRF SaMD risk categorization matrix (IMDRF/SaMD WG/N12FINAL:2014) uses two axes, significance of information to healthcare decisions and state of the healthcare condition, to assign SaMD into four risk categories (I–IV). These categories inform FDA classification and submission pathways. The table below maps each category to a cardiology-specific example.
| IMDRF Risk Category | Significance of Information | Healthcare Condition State | Cardiology Example | Typical FDA Class |
|---|---|---|---|---|
| I (Lowest) | Inform clinical management | Non-serious | General wellness heart-rate trending app | Class I / Exempt |
| II | Inform clinical management | Serious | RPM dashboard displaying HF weight trends to a clinician | Class II (510(k)) |
| III | Drive clinical management | Non-serious | CDS tool recommending medication titration for hypertension | Class II (510(k) or De Novo) |
| IV (Highest) | Drive clinical management | Critical/life-threatening | AI algorithm autonomously detecting VF and triggering therapy recommendations | Class III (PMA) |
| II–III (Hybrid) | Diagnose/detect | Serious | AI-based AFib detection from CIED transmission data | Class II (De Novo or 510(k)) |
Cardiology accounts for roughly 10% of all FDA-cleared AI-enabled medical devices, ranking second to radiology. Approximately 97% of cleared AI/ML devices have used the 510(k) pathway, which makes this route the dominant option for cardiac remote monitoring platforms.
Understanding how the FDA classifies cardiac monitoring software by risk is only half of the picture. The other half is determining whether a given piece of software is regulated as a standalone device (SaMD) or as part of a hardware device (SiMD).
Software in a Medical Device (SiMD) is software embedded within or necessary for the operation of a physical medical device. The hardware cannot achieve its medical purpose without the software, and SiMD is regulated as part of the parent device rather than as a standalone product.
In a CIED ecosystem, the distinction is concrete.
Hybrid products such as cloud-connected cardiac monitoring systems commonly contain both SiMD components and SaMD components. Each component requires separate classification and documentation.
The IMDRF two-question classification test provides a practical lens. First, does the software meet the definition of a medical device based on intended purpose. Second, is the software necessary for a hardware device to achieve its medical purpose (SiMD), or can it function independently on general-purpose platforms (SaMD). A vendor-neutral CIED data platform that runs on cloud infrastructure and performs independent analysis answers “no” to the second question, so it is SaMD.
Several interlocking regulatory instruments govern RPM software used in cardiology as of July 2026, and together they define expectations for both manufacturers and clinical buyers.
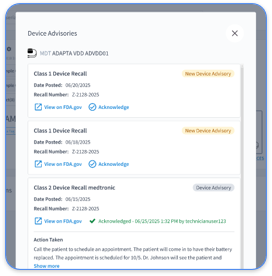
Multi-vendor cardiac platforms routinely incorporate off-the-shelf (OTS) software components such as commercial databases, cloud infrastructure services, HL7 parsing libraries, and third-party AI frameworks. Software of Unknown Provenance (SOUP) incorporated into cardiac remote monitoring platforms must have its failure modes included in the ISO 14971 hazard analysis and addressed within the IEC 62304 risk management and verification processes.
For a platform ingesting data from Medtronic, Boston Scientific, Abbott, and Biotronik portals simultaneously, each OTS parsing component, third-party API wrapper, and cloud service layer requires a documented SOUP inventory, functional requirements specification, and verification testing in the specific device context. FDA inspections of SaMD manufacturers frequently identify missing or incomplete SOUP inventories.
As of February 2, 2026, the FDA’s QMSR replaced the legacy Quality System Regulation (21 CFR 820) by formally incorporating ISO 13485:2016 by reference, enabling SaMD manufacturers to leverage the same standard for both FDA and international compliance.
For cardiology remote monitoring platforms, ISO 13485 compliance requires documented processes that span the full product lifecycle.
On December 4, 2024, the FDA published final guidance titled "Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions," finalizing the PCCP framework for AI-enabled medical devices. The final PCCP guidance for AI-enabled devices was issued by the FDA in December 2024.
A PCCP submitted with a 510(k), De Novo, or PMA application must include three components.
The FDA’s draft Total Product Life Cycle (TPLC) guidance for AI-enabled device software functions, issued January 7, 2025, remains unfinalized as of June 2026. It proposes submission elements including model description, data lineage, bias analysis, human-AI workflow, and real-world performance monitoring.
Approximately 10% of 2025 AI/ML clearances included PCCPs for future algorithm updates, which signals growing adoption and also shows that many vendors still operate without this flexibility. For cardiac AI tools such as AFib detection algorithms or arrhythmia triage models, a PCCP is the mechanism that allows iterative model improvement without a new 510(k) submission for each update.
Every cardiac remote monitoring SaMD must achieve FDA clearance, and for most AI/ML devices that means a 510(k) submission. Understanding the core elements of that submission helps administrators evaluate vendor claims and verify compliance.
A 510(k) submission for a cardiac remote monitoring SaMD platform requires the following core documentation elements.
Practices managing patients across multiple CIED manufacturers encounter recurring compliance and operational failures that often compound each other.
University of Chicago Medicine reviewed more than 73,000 reports annually through Rhythm360 in calendar year 2025, averaging more than 18,000 reports per quarter. This volume demonstrates the platform’s capacity in high-volume cardiology environments. Clinicians at UCM reported being able to "address these issues earlier; rather than waiting for a 3-month visit, we can call patients in for evaluation."
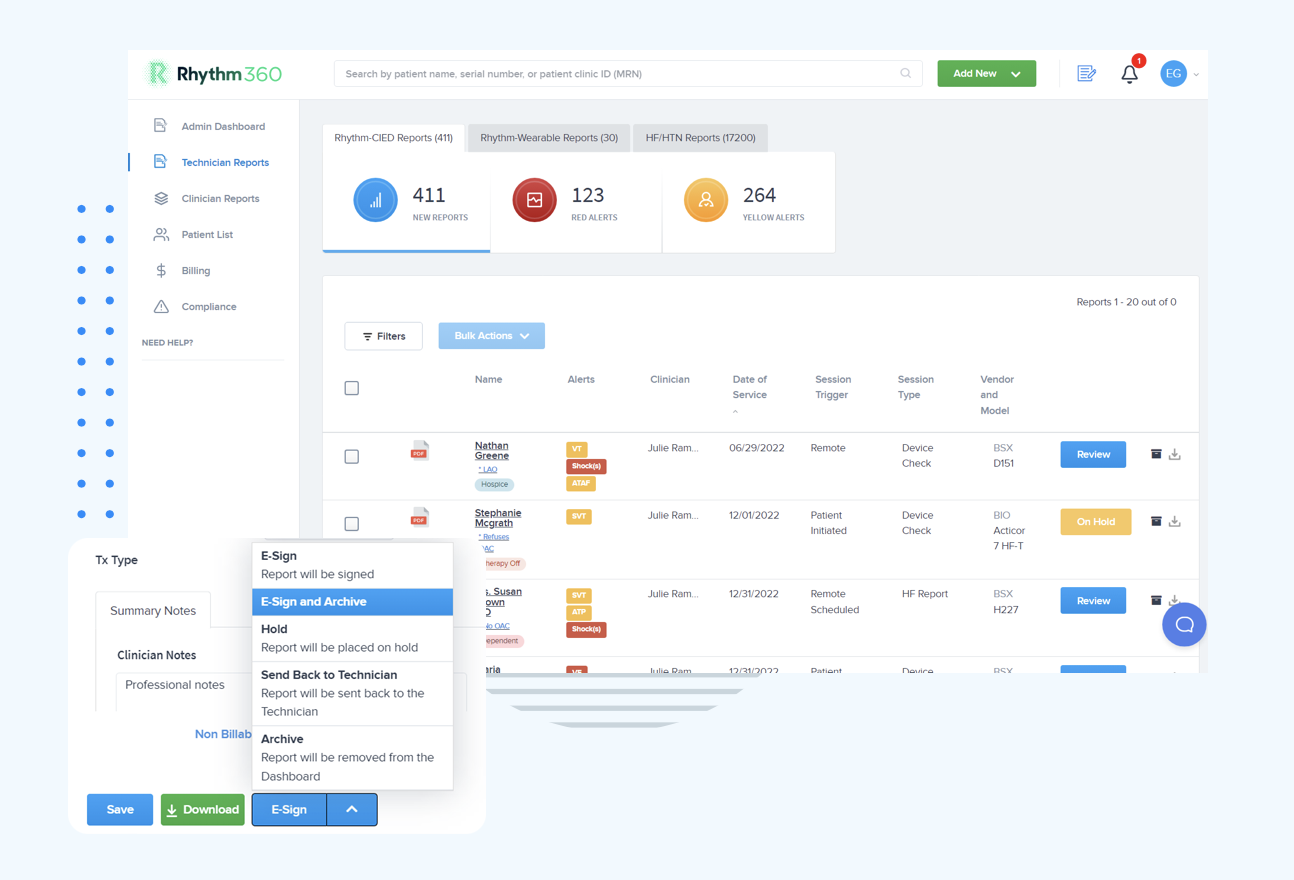
The table below maps Rhythm360’s core capabilities to clinical outcomes and regulatory expectations so administrators can see how specific features support compliance.
| Rhythm360 Capability | How It Works | Clinical/Operational Outcome | Compliance Relevance |
|---|---|---|---|
| Vendor-neutral CIED data ingestion | API, HL7, XML, and PDF parsing via computer vision (OCR) from Medtronic, Boston Scientific, Abbott, Biotronik, and others | Single dashboard eliminates multi-portal logins, and greater than 99.9% transmissibility via redundant data feeds | Supports SOUP documentation and data integrity requirements under IEC 62304 and ISO 14971 |
| AI-powered alert triage | AI-driven prioritization filters non-actionable transmissions and surfaces clinically significant events in near-real-time | Up to 80% reduction in critical alert response times with optional 24/7/365 CCT oversight | Aligns with GMLP transparency and bias-mitigation expectations in FDA’s January 2025 draft TPLC guidance |
| Automated CPT billing documentation | Automated report generation with full audit trail for billable remote monitoring events | Practices have achieved up to 300% increase in revenue through improved CPT code capture | Auditable documentation supports post-market surveillance and complaint-handling requirements |
| Bi-directional EHR integration | Native integrations with Epic, Cerner, Athenahealth, eClinicalWorks, and Greenway Health via HL7 | Eliminates manual data transcription and enables onboarding in days to weeks | Supports design control and change control documentation under QMSR and ISO 13485 |
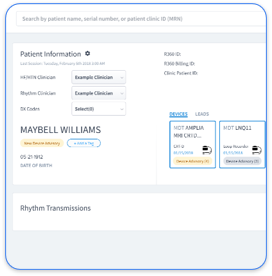
Yes, in most cases. A platform that ingests CIED transmission data and performs independent analysis, generates clinical alerts, or provides decision support qualifies as SaMD under the IMDRF definition adopted by the FDA. The fact that the underlying hardware, the implanted device, is separately cleared does not exempt the software platform from its own regulatory obligations. Practices should request the FDA clearance number and intended use documentation from any software vendor before deployment. Rhythm360 is a HIPAA-compliant platform built to meet current FDA expectations for cardiac remote monitoring SaMD.
IEC 62304 classifies software by the severity of harm that could result from a software failure. Class B applies where failure could cause non-serious injury. Class C applies where failure could cause death or serious injury. For cardiac remote monitoring software, alert triage algorithms that detect ventricular fibrillation, lead malfunction, or critical arrhythmias are typically classified as Class C because a missed or delayed alert could directly contribute to patient death or serious harm. Class C requires comprehensive verification, detailed architectural design documentation, unit testing for every software unit, and full bidirectional traceability from requirements through risk controls to test cases. Practices evaluating software vendors should ask for the vendor’s IEC 62304 safety class determination and corresponding V&V documentation.
A Predetermined Change Control Plan (PCCP) is a document submitted with a device’s initial 510(k), De Novo, or PMA application that pre-authorizes specific future modifications to an AI algorithm, such as retraining on new patient data or adjusting detection thresholds, without requiring a new submission for each change. The FDA finalized the PCCP framework in December 2024 and updated it in August 2025. For cardiology practices, the practical implication is that a vendor whose AI alert triage model operates under an authorized PCCP can update that model within defined bounds without regulatory delay. Practices should ask vendors whether their AI features are covered by an authorized PCCP and what the scope of covered modifications includes.
The FDA’s Quality Management System Regulation (QMSR), effective February 2, 2026, replaces the legacy 21 CFR Part 820 Quality System Regulation and formally incorporates ISO 13485:2016 by reference. For SaMD manufacturers, including cardiac remote monitoring platform vendors, this shift means design controls, risk management, supplier oversight, and post-market surveillance must now align with ISO 13485 requirements. Practices should confirm that their software vendors have updated their quality management systems to QMSR compliance and can provide evidence of ISO 13485 certification or equivalent documentation. Vendors who have not transitioned may be operating outside current FDA expectations.
Yes. Rhythm360 is purpose-built for multi-OEM environments. The platform ingests and normalizes data from Medtronic, Boston Scientific, Abbott, Biotronik, and other manufacturers through a combination of API connections, HL7 feeds, XML parsing, and AI-powered computer vision for PDF-based transmissions. A redundant data feed architecture maintains greater than 99.9% transmissibility even when an individual OEM server is unavailable. Device technicians and electrophysiologists access a single unified dashboard rather than logging into separate manufacturer portals, and the platform’s automated reporting generates auditable documentation for CPT billing across the full CIED patient population.
The FDA’s regulatory framework for medical device software has matured significantly between 2024 and 2026. The QMSR’s alignment with ISO 13485, the finalized PCCP framework for AI-enabled devices, mandatory cybersecurity premarket documentation, and the January 2025 draft TPLC guidance collectively raise the compliance bar for any software platform used in cardiac remote monitoring. Practices that adopt platforms without verifying FDA clearance status, PCCP coverage, IEC 62304 safety classification, and QMSR compliance accept both regulatory and clinical risk.
Rhythm360 is designed to meet these expectations while addressing the operational problems that fragmented multi-OEM workflows create, including alert fatigue, data silos, missed critical events, and billing documentation gaps. The platform’s track record includes supporting more than 73,000 annual reports at a single academic medical center and delivering up to 80% reductions in critical alert response times across its user base.